
Antimitochondrial Antibody–Negative Primary Biliary Cirrhosis in an Elderly African American Man
Primary biliary cirrhosis (PBC) is a chronic, progressive autoimmune liver disease that is characterized by nonsuppurative destruction of intrahepatic ducts, resulting in cholestasis, cirrhosis, and end-stage liver disease.1,2 Antimitochondrial antibodies (AMAs) are considered to be a highly sensitive and specific serological marker of PBC; however, a subset of patients who have biochemical and histopathological features consistent with PBC do not have detectable AMAs in their serum.1-5 This phenomenon is referred to as AMA-negative PBC. Although AMA-negative PBC is predominantly diagnosed in middle-aged Caucasian women, it has been observed on occasion in elderly men, especially those of African American descent.6 We report one such case in a 67-year-old African American man and provide a brief review of the literature on AMA-negative PBC.
Case Report
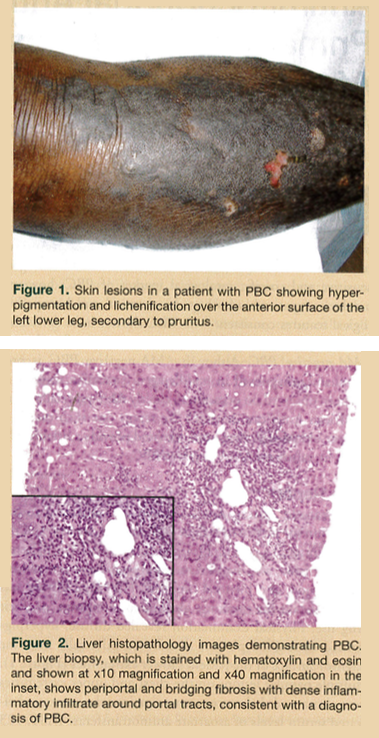
A 67-year-old African American man was admitted to our institution for increased fatigue, generalized itching, and the gradual onset of a rash on his lower extremities. The patient’s medical history was significant for type 2 diabetes mellitus, hypertension, and bilateral cataracts. His social history was not significant for smoking, alcohol consumption, or illicit drug use. Physical examination revealed diffuse, hyperpigmented lichenified plaques distributed bilaterally over the extensor surface of the patient’s lower extremities (Figure 1). Multiple papular lesions on the chest and upper extremities were also noted. Laboratory investigations demonstrated elevated serum levels of aspartate aminotransferase (AST, 123 U/L; normal, 10-30 U/L), alanine aminotransferase (ALT, 125 U/L; normal, 10-40 U/L), alkaline phosphatase (ALP, 930 U/L; normal, 30-120 U/L), total bilirubin (2.9 mg/dL; normal, 0.3-1.2 mg/dL), and direct bilirubin (2.1 mg/dL; normal, 0.1-0.3 mg/dL). Serology for antihepatitis B surface antibody, antihepatitis C antibody, antinuclear antibody (ANA), and AMA were negative. Abdominal ultrasonography showed no gallstones, but there was increased echogenicity throughout the liver, consistent with diffuse hepatocellular disease. A subsequent liver biopsy demonstrated histological features consistent with stage 3 PBC with periportal and bridging fibrosis, but no cirrhosis (Figure 2). The patient was treated with oral ursodeoxycholic acid. Periodic liver function tests over the next 6 years revealed marked improvements in the patient’s AST (74 U/L), ALT (58 U/L), ALP (172 U/L), and total and direct bilirubin levels (1.5 mg/dL and 0.9 mg/dL, respectively).
Demographics of Primary Biliary Cirrhosis
PBC has been considered a disease of middle-aged Caucasian women, generally occurring between the ages of 40 and 65 years.6 Studies have shown a strong female to male preponderance, ranging from 6:1 to 22:1.7,8 Because most patients with PBC are of Caucasian ethnicity, data on the clinical presentation and disease severity in other ethnic groups are limited, especially with regard to older persons.
A large, prospective, randomized, US-based multicenter trial conducted by Peters and colleagues6 that included 535 patients with PBC showed a similar mean age (52.1 ± 9.5 years vs 51.5 ± 9.4 years), female to male predominance (13.9:1 vs 12.2:1), and AMA-positive rates (92.4% vs 86.3%) between Caucasian and non-Caucasian (African American and Hispanic) populations, respectively; however, the majority of patients (86.4%) in this study were Caucasian. Results from this cohort study suggest a greater prevalence of PBC in Caucasians than in non-Caucasian populations, but a major limitation of this study, as acknowledged by its authors, was that most of the participants were enrolled from tertiary referral centers and did not represent the actual demographics of the general population. The study did show a trend toward greater disease severity in the non-Caucasians, who generally also demonstrated lower activity levels, more severe pruritus, and more advanced disease than Caucasian patients, resulting in more individuals in this group being excluded from the clinical trial (46.5% vs 25.1%, respectively).6
According to a Canadian study, the age-adjusted incidence rates of PBC observed among patients aged 60 to 79 years and those aged 80 years and older were 63 and 22.4 per 1 million, respectively, and the adjusted point prevalence rates were 573 and 137 per 1 million, respectively.9 In women aged 40 to 59 years, the adjusted incidence and point prevalence rates were 59 and 437 per 1 million, respectively. A limitation of this study is that it did not address racial disparities in the age- and sex-adjusted incidence and point prevalence rates of PBC.9 Although PBC has been reported in all ethnic groups, the exact demographic data on the incidence and prevalence of PBC among African Americans and other ethnic groups compared with Caucasians, especially among individuals older than 65 years, is still unknown.
Environmental and Genetic Factors
Environmental and genetic factors have been shown to influence susceptibility to PBC. Currently, there is evidence to support the role of infectious agents, such as bacteria, retroviruses, and xenobiotics, in the pathogenesis of PBC.10,11 It is thought that exposure to these agents, particularly xenobiotics, can alter the structure of native proteins, inducing an immune response to self-proteins, a phenomenon referred to as molecular mimicry.10 Identification of several PBC clusters lends anecdotal support to this theory, as multiple cases of PBC have been reported within a single family, both among genetically related individuals and genetically unrelated individuals living in the same household.12 In addition, a high prevalence of PBC has been observed in certain geographic areas.12,13 One study by investigators from the United Kingdom and Greece reported that PBC clusters have often been identified in areas close to water reservoirs, coal mining areas, and toxic waste disposal sites, adding strength to the notion that environmental factors may serve as a catalyst to PBC.13
Several studies have identified a significant risk among persons genetically related to patients with PBC compared with the risk noted in the general population.14,15 Studies examining the disease association with major histocompatibility complex class II molecules have identified HLA-DR8, DRB1*08, DR3, DPB1*0301, DR2, DPB1*0501, and DRB1*0803 as risk factors for PBC.7 Significant genetic associations with PBC include polymorphisms in the tumor necrosis factor-a promoter region, the TAP1/TAP2 genes on chromosome 6, the promoter region of the IL-10 gene, the natural resistance-associated macrophage protein 1 gene, and the vitamin D receptor gene.7
Diagnosis
According to the American Association for the Study of Liver Diseases (AASLD) guidelines, PBC should be considered in patients with elevated serum ALP levels and the diagnosis established if two of the following three criteria are met: AMA is detected; elevated ALP levels are indicative of cholestasis; and a liver histology confirms the nonsuppurative destruction of intrahepatic ducts.16 With the sensitivity and specificity of AMAs approaching 95%, detection of AMA is important in the diagnosis of PBC.1 It is estimated that as many as 84% to 97% of patients with PBC demonstrate positive AMA titers; however, a small proportion of patients with PBC have no AMAs discernable in their serum, and this subset is referred to as having AMA-negative PBC.1-5 Indirect immunofluorescence is presently used for the detection of AMAs in many laboratories. Up to 15% of patients with PBC may be identified as AMA-negative by this method.17 Novel techniques, such as enzyme-linked immunosorbent assays (ELISA) and immunoblotting, have been useful in detecting AMAs in up to 79% of patients with AMA-negative PBC.17,18
A higher titer of ANA, anti-smooth muscle antibody, gamma globulin, and lower immunoglobulin M (IgM) have been demonstrated in patients with AMA-negative PBC compared with those with AMA-positive PBC4; however, these disease entities have been shown to have comparable biochemical and histological features and a similar clinical course.5 Liu and colleagues5 suggested that AMA-negative PBC may not truly be negative, but that it may represent an atypical variant of AMA-positive PBC, rather than a separate clinical entity. In this study, which included 12 patients with AMA-negative PBC and 12 with AMA-positive PBC, assessment of patients’ clinical features, liver biochemistries and histology, and immunological parameters, including anti-glycoprotein-210, anti-Sp100, and CD4+ CD25+ regulatory T cells, revealed no significant difference between those with AMA-negative and those with AMA-positive PBC. Although higher titers of ANA were observed in patients with AMA-positive PBC, this finding did not reach statistical significance, as the sample size was small (n=12).5
Treatment
Once a diagnosis of PBC is established, regardless of whether it is positive or negative for AMAs, the most widely accepted treatment is ursodeoxycholic acid. This pharmacotherapy is the only one approved by the US Food and Drug Administration (FDA) as a treatment for PBC and the only agent endorsed by the AASLD for this purpose.16 Ursodeoxycholic acid is well tolerated in most patients. The most commonly reported side effects are headache, constipation, and diarrhea.19
In Liu and colleagues’ study,5 treatment of AMA-negative and AMA-positive PBC with ursodeoxycholic acid resulted in similar improvements in ALP, IgM, and glutamyl transpeptidase levels. Both groups also showed a significant increase of regulatory T cells after a 1-year follow-up.In a retrospective analysis by Mayo Clinic researchers, the biochemical parameters and Mayo risk score for patients with AMA-negative and AMA-positive PBC were similar, and both groups demonstrated similar treatment outcomes with ursodeoxycholic acid.20 As noted in our case report, serial monitoring of our patient’s liver function over a 6-year period demonstrated significant improvements in his AST, ALT, ALP, and total and direct bilirubin levels following initiation of usodeoxycholic acid.
Other pharmacotherapies that have been used to treat PBC include colchicine and methotrexate, but these have been prescribed off-label, as their role is controversial and their benefits are less clear.21 Therefore, once ursodeoxycholic acid ceases to control the disease and the patient progresses to end-stage liver disease, liver transplantation should be considered, regardless of the patient’s AMA status.22 In a study by Kim and colleagues,23 the clinical outcomes following orthotopic liver transplantation in patients with AMA-negative PBC were examined after a median follow-up of 36-months. The authors concluded that the graft and patient survival rates and subsequent histological changes, including disease recurrence and steroid-resistant or late rejections, were comparable with those of patients with AMA-positive PBC.23
Conclusion
AMA-negative and AMA-positive PBC share similar clinical, biochemical, and histopathological features. Improvement in sensitivity of techniques for detecting AMAs will further bridge the gap between AMA-negative and AMA-positive PBC. Therefore, detection of AMAs alone should not influence the diagnosis or treatment of PBC, and the disease should be managed in accordance with the AASLD guidelines.16
The authors report no relevant financial relationships.
References
1. Kaplan MM, Gershwin ME. Primary biliary cirrhosis. N Engl J Med. 2005;353(12):1261-1273.
2. Selmi C, Invernizzi P, Zuin M, Podda M, Seldin MF, Gershwin ME. Genes and (auto)immunity in primary biliary cirrhosis. Genes Immun. 2005;6(7):543-556.
3. Bassendine MF, Yeaman SJ. Serological markers of primary biliary cirrhosis: diagnosis, prognosis and subsets. Hepatology.1992;15(3):545-548.
4. Michieletti P, Wanless IR, Katz A, et al. Antimitochondrial antibody negative primary biliary cirrhosis: a distinct syndrome of autoimmune cholangitis. Gut. 1994;35(2):260-265.
5. Liu B, Shi XH, Zhang FC, Zhang W, Gao LX. Antimitochondrial antibody-negative primary biliary cirrhosis: a subset of primary biliary cirrhosis. Liver Int. 2008;28(2):233-239.
6. Peters MG, Di Bisceglie AM, Kowdley KV, et al; for the PUMPS Group. Differences between Caucasian, African American, and Hispanic patients with primary biliary cirrhosis in the United States. Hepatology. 2007;46(3):769-775.
7. Feld JJ, Heathcote EJ. Epidemiology of autoimmune liver disease. J Gastroenterol Hepatol. 2003;18(10):1118-1128.
8. Triger DR, Berg PA, Rodes J. Epidemiology of primary biliary cirrhosis. Liver. 1984;4(3):195-200.
9. Myers RP, Shaheen AA, Fong A, et al. Epidemiology and natural history of primary biliary cirrhosis in a Canadian health region: a population-based study. Hepatology. 2009;50(6):1884-1892.
10. Van de Water J, Ishibashi H, Coppel RL, Gershwin ME. Molecular mimicry and primary biliary cirrhosis: premises not promises. Hepatology. 2001;33(4):771-775.
11. Long SA, van de Water J, Gershwin ME. Antimitochondrial antibodies in primary biliary cirrhosis: the role of xenobiotics. Autoimmun Rev. 2002;1(1-2):37-42.
12. Abu-Mouch S, Selmi C, Benson GD, et al. Geographic clusters of primary biliary cirrhosis. Clin Dev Immunol. 2003;10(2-4):127-131.
13. Smyk D, Mytilinaiou MG, Rigopoulou EI, Bogdanos DP. PBC triggers in water reservoirs, coal mining areas and waste disposal sites: from Newcastle to New York. Dis Markers. 2010;29(6):337-344.
14. Kim WR, Lindor KD, Locke GR, et al. Epidemiology and natural history of primary biliary cirrhosis in a US community. Gastroenterology. 2000;119(6):1631-1636.
15. James OFW, Bhopal R, Howl D, Gray J, Burt AD, Metcalf JV. Primary biliary cirrhosis once rare, now common in the United Kingdom? Hepatology. 1999;30(2):390-394.
16. Lindor KD, Gershwin ME, Poupon R, et al; American Association for Study of Liver Diseases. Primary biliary cirrhosis. Hepatology. 2009;50(1):291-308.
17. Kitami N, Komada T, Ishii H, et al. Immunological study of anti-M2 in antimitochondrial antibody-negative primary biliary cirrhosis. Intern Med. 1995;34(6):496-501.
18. Nakajima M, Shimizu H, Miyazaki A, et al. Detection of IgA, IgM, and IgG subclasses of anti-M2 antibody by immunoblotting in autoimmune cholangitis: is autoimmune cholangitis an early stage of primary biliary cirrhosis? J Gastroenterol. 1999;34(5):607-612.
19. Siegel JL, Jorgensen R, Angulo P, Lindor KD. Treatment with ursodeoxycholic acid is associated with weight gain in patients with primary biliary cirrhosis. J Clin Gastroenterol. 2003;37(2):183-185.
20. Lacerda MA, Ludwig J, Dickson ER, Jorgensen RA, Lindor KD. Antimitochondrialantibody-negative primary biliary cirrhosis. Am J Gastroenterol. 1995;90(2):247-249.
21. Kaplan MM, Cheng S, Price LL, Bonis PA. A randomized controlled trial of colchicine plus ursodiol versus methotrexate plus ursodiol in primary biliary cirrhosis: ten-year results. Hepatology. 2004;39(4):915-923.
22. Lee J, Belanger A, Doucette JT, et al. Transplantation trends in primary biliary cirrhosis. Clin Gastroenterol Hepatol. 2007;5(11):1313-1315.
23. Kim WR, Poterucha JJ, Jorgensen RA, et al. Does antimitochondrial antibody status affect response to treatment in patients with primary biliary cirrhosis? Outcomes of ursodeoxycholic acid therapy and liver transplantation. Hepatology. 1997;26(1):22-26.