
A Child With Bone and Craniofacial Abnormalities
A 1-day-old girl born to a 24-year-old gravida 2, para 1 mother at almost 37 weeks’ gestation was referred to our facility with concern regarding a soft skull. Routine prenatal ultrasonography results had shown underdeveloped skull bones. The mother’s past medical history was significant for surgically treated bilateral hip dysplasia and significantly shorter stature than her siblings. The neonate’s maternal grandmother had extra teeth. The mother had an elective abortion of her first pregnancy when the fetus was diagnosed with renal agenesis and Potter sequence.
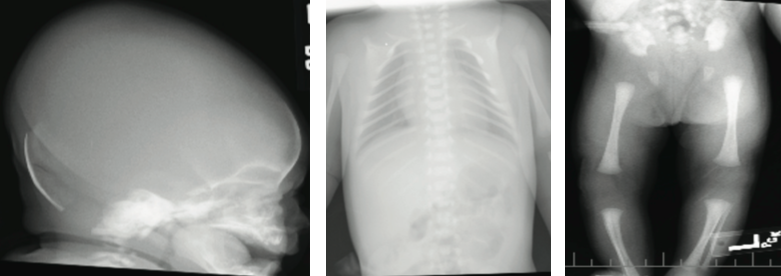
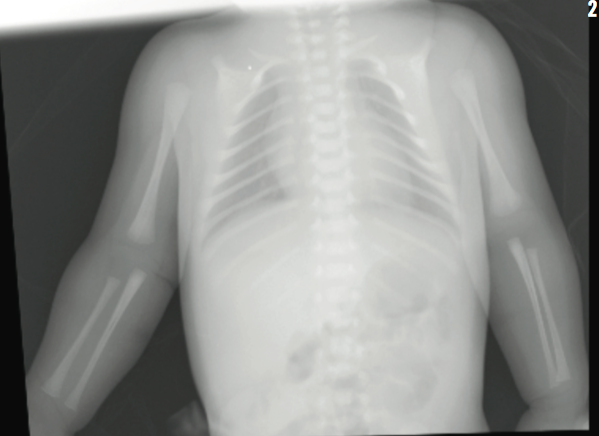
The infant was born via spontaneous vaginal delivery. Apgar scores were normal. Weight, length, and head circumference were appropriate for gestational age. Her initial newborn examination was notable for large anterior fontanelle (7 cm in transverse diameter), wide metopic suture, craniotabes, broad nasal bridge, and bilateral fifth digit clinodactyly. Examination of posterior sutures was difficult due to caput succedaneum. Bilateral clavicles were palpated medially, but not laterally.
The results of transfontanelle ultrasonography of the head were unremarkable, and radiography of the skull revealed thinning of the calvarium, frontal bossing, and increased anteroposterior diameter of the skull. Calcium, phosphorus, alkaline phosphatase, and vitamin D serum levels were within reference ranges. Skeletal survey revealed absence of the lateral two-thirds of each clavicle, nonossification of pubis, and unremarkable positioning of the hip joints. The neonate was discharged home on day 4 of life.
The girl is now 2 years old. Her skull bones are still underdeveloped but are progressively getting better. Her height is at the 50th percentile for her age. Her dentition is appropriate for her age, and her repeated hearing tests all have been normal. She has developed progressive infantile scoliosis for which she is being managed with serial casting.
Genetic workup will be consistent with the diagnosis of?
A. Holt-Oram syndrome
B. Cleidocranial dysplasia
C. Pyknodysostosis
D. Osteogenesis imperfecta
Answer: Cleidocranial dysplasia
Genetic testing included targeted DNA sequencing of the runt-related transcription factor 2 (RUNX2) gene and high-resolution chromosome (microarray) analysis. Frameshift mutation in the RUNX2 gene was consistent with the diagnosis of cleidocranial dysplasia.
Discussion
Cleidocranial dysplasia, also known as cleidocranial dysostosis, is a rare hereditary disorder inherited in an autosomal dominant pattern. It is characterized by defective ossification, delayed bone and tooth development, stomatognathic abnormalities, and craniofacial abnormalities. The disorder is caused by mutations in the RUNX2 gene that is responsible for osteoblast differentiation.1,2 More than 70 mutations of the RUNX2 gene have been described in individuals with cleidocranial dysplasia.3,4 The RUNX2 mutation controls the activity of genes involved in the development and maintenance of bone and cartilage. Mutations may be hereditary, although 40% of cleidocranial dysplasia cases appear spontaneously with no apparent genetic cause.5 As an autosomal dominant condition, an affected individual has a 50% chance of passing on the disorder to offspring.
Radiographic findings of cleidocranial dysplasia include delay in closure of the skull fontanelles, hypertelorism, partial or complete absence of clavicles, and lack of ossification of the pubis.2,6 None of the primary clinical findings have serious sequelae.
Individuals with cleidocranial dysplasia may demonstrate a spectrum of clinical findings that have varying severity. Associated clinical findings include short stature, scoliosis, ligamentous laxity, hearing impairment, dental anomalies in both primary and secondary teeth, and osteoporosis.6,7 The majority of affected individuals remain asymptomatic throughout life with only a minority developing upper extremity symptoms due to stress imposed on the brachial plexus. Physical therapy frequently resolves the symptoms, and surgical intervention is rarely required.8 Additionally, Cooper and colleagues9 found no evidence showing statistically significant delays in developmental, intellectual, and motor skills in patients with cleidocranial dysplasias compared with controls.
The differential diagnosis of a large fontanelle with associated skeletal problems includes more common disorders such hydrocephalus (primary or associated with Dandy-Walker syndrome and Chiari malformations), rickets, or hypothyroidism. Skeletal dysplasias include osteogenesis imperfecta, achondroplasia, hypophosphatasia, as well as cleidocranial dysplasias.10 Congenital pseudarthrosis is a rare cause of clavicular hypoplasia affecting the middle and lateral thirds of the bone, always on the right side, and lacking significant family history or other skeletal anomalies.11,12
Because early diagnosis of cleidocranial dysplasia is essential for initiating appropriate treatment, clinicians should be aware of the characteristic symptoms.13 Given the complications of skeletal dysplasias like cleidocranial dysplasia, it is important for clinicians to have a high index of suspicion for skull and skeletal changes in infants so appropriate imaging studies are obtained in the newborn period. Early diagnosis of cleidocranial dysplasia has proven benefit. A multidisciplinary approach is recommended to manage these patients including regular dental appointments, orthopedic follow-up, and hearing tests. n
Muhammad Ubaidulhaq, MD, is from the department of child neurology at Wayne State University, and Children’s Hospital of Michigan, both in Detroit.
Alyssa Bottrell, MS, is an MD/PhD student at Wayne State University in Detroit, Michigan.
Safwan Riaz, MD, is a research associate at Children’s Hospital of Michigan in Detroit.
References
1. D’Alessandro G, Tagariello T, Piana G. Cleidocranial dysplasia: etiology and stomatognathic and craniofacial abnormalities. Minerva Stomatol. 2010;59(3):117-27.
2. Al Kaissi A, Ben Chehida F, Kenis V, et al. Broad spectrum of skeletal malformation complex in patients with cleidocranial dysplasia syndrome: radiographic and tomographic study. Clin Med Insights Arthritis Musculoskelet Disord. 2013;19(6):45-55.
3. Pawłowska E, Wójcik KA, Synowiec E, Szczepańska J, Błasiak J. Expression of RUNX2 and its signaling partners TCF7, FGFR1/2 in cleidocranial dysplasia. Acta Biochim Pol. 2015;62(1):123-126.
4. Cohen MM Jr. Perspectives on RUNX genes: an update. Am J Med Genet A. 2009;149A(12):2629-2646.
5. Tanaka JL, Ono E, Filho EM, Castilho JC, Moraes LC, Moraes ME. Cleidocranial dysplasia: importance of radiographic images in diagnosis of the condition. J Oral Sci. 2006;48(3):161-166.
6. Candamourty R, Venkatachalam S, Yuvaraj V, Kumar GS. Cleidocranial dysplasia with hearing loss. J Nat Sci Biol Med. 2013;4(1):245-249.
7. Quack I, Vonderstrass B, Stock M, et al. Mutation analysis of core binding factor A1 in patients with cleidocranial dysplasia. Am J Hum Genet. 1999;65(5):1268-1278.
8. Morcuende JA, Dobbs MB. Idiopathic and heritable disorders; defects in nuclear proteins and transcription factors; cleidocranial dysplasia. In: Weinstein SL, Buckwalter JA, eds. Turek’s Orthopaedics: Principles and Their Application. 6th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2005: 252.
9. Cooper SC, Flaitz CM, Johnston DA, Lee B, Hecht JT. A natural history of cleidocranial dysplasia. Am J Med Genet. 2001;104(1):1-6.
10. Carlo WA. Part XII; The fetus and the neonatal infant: The newborn infant. In: Kliegman RM, Stanton BF, St Geme JW, Schor NF, Behrman RE, eds. Nelson Textbook of Pediatrics. Vol.16.20th ed. Philadelphia, PA: Elsevier; 2016:795.
11. Cadilhac C, Fenoll B, Peretti A, Padovani JP, Pouliquen JC, Rigault P. Congenital pseudarthrosis of the clavicle: 25 childhood cases. Rev Chir Orthop Reparatrice Appar Mot. 2000;86(6):575-580.
12. Alldred AJ. Congenital pseudarthrosis of the clavicle. J Bone Joint Surg Br. 1963;45-B:312-319.
13. Golan I, Baumert U, Hrala BP, Müssig D. Dentomaxillofacial variability of cleidocranial dysplasia: clinicoradiological presentation and systematic review. Dentomaxillofac Radiol. 2003;32(6):347-354.