Neuromuscular Diseases in Geriatric Patients: Part I
Most biologic capabilities decline with age, although the extent of the decrements are usually modest and vary by function. Cross-sectional studies show that strength declines by approximately 30% from the third through eighth decades and that sensory changes, while difficult to measure, are comparable.1 The effects of disease are superimposed on age-related decreases often resulting in significant functional impairment. Many neuromuscular diseases are uncommon in older persons. This two-part article will focus on the conditions that occur with regularity in older persons or have a striking effect on their lives. Part I discusses cervical spondylotic radiculomyelopathy, lumbar spondylosis, amyotrophic lateral sclerosis (ALS), Guillain-Barré syndrome (GBS), and acquired demyelinating polyneuropathies. Part II, which will be published in the next issue of the Journal, will focus on myasthenia gravis, inclusion body myositis, polymyositis, and polymyalgia rheumatica.
The signature manifestation of neuromuscular disease is weakness with a loss of the function that it supports. Lower-extremity weakness impairs mobility, while weakness of the upper extremities may compromise activities of daily living (ADL) or instrumental activities of daily living (IADL), requiring use of the arms and/or hands. Evaluation of strength with manual motor testing in older persons is problematic for inexperienced clinicians because of age-related strength decrements and as a consequence of variability due to activity level and gender. To assist in interpretation of manual motor testing, comparison of muscles may serve as a framework for loss of strength in specific muscles (eg, right against left or distal vs proximal). Due to these problems, functional testing of the motor system is useful for assessing strength. Functional testing should include the following:
1. Arising from chair without use of arms. Lift-off requires strength of knee and hip extensors, while balance is required to control center of mass as it moves forward (about 24 in) from sitting position to being supported by lower extremities. If patient cannot arise, then allow use of arms to determine if lift (proximal strength), steadying (balance), or both are provided by upper extremities. Examiner may either hold both hands to determine role of upper extremity or estimate force exerted on chair’s arms. Weakness can be confirmed by manual motor testing of glutei and quadriceps, while balance will be tested by other functional testing.
2. Performing one- or two-leg heel and toe rises. Depending on age and frailty, this requires dorsi and plantar extension strength. Provide support by holding both of patient’s hands. Ask patient to lift (suspend) one leg and while providing support by holding both hands, ask him/her to lean forward, lifting up onto the toes, and then rocking backward onto the heel, lifting forefoot from ground. Perform on other lower extremity. If unable to perform on one leg, have the patient do it on both. Inability to go up onto toes indicates weakness of plantar flexion (gastrocnemius), while failure to rock backward onto heels suggests weakness of dorsiflexors (tibialis anterior). The extent of steadying required through support of the arms provides some indication of balance.
3. Performing single, tandem, or semi-tandem stance. This requires strength but primarily tests balance. Single or tandem (toe-to-toe) stance time for a healthy 80-year-old is 10 seconds or more, with lower times suggesting mild balance impairment. Ability to maintain balance only with feet together (side-by-side) suggests moderate impairment, and inability to maintain this stance indicates a more severe problem.
4. Testing balance using forward, backward, and side pulls. Examiner should pull patient toward him/herself to use his/her body to check motion before patient loses control of his/her body mass. There is a larger zone of stability anteriorly, making forward pulls easier to control by comparison with those to the rear. A normal patient should be able to control moderate pull with body sway. The use of 1-2 steps suggests mild balance impairment. Incipient loss of balance requiring examiner intervention suggests moderate or severe impairment, depending on extent of loss of control.
5. Testing gait movements. Although gait velocity of a normal older person may slow modestly, gait should remain a series of coordinated movements of upper and lower extremity with repetitive heelstrikes and toe-offs. Posture should be upright. Turns (180 degrees) should occur with one to three steps. Corticospinal disease produces stiff gait with scissoring (feet touch). Foot dorsiflexor weakness (footdrop) produces high stepping with tendency to catch toes. Plantar flexor weakness produces limp with inability to toe-off, while proximal weakness causes waddling gait. A severe sensory deficit due to neuropathy may produce mild ataxia (wide-base with unsteadiness), which worsens in the dark or produces unsteadiness when standing with eyes closed and feet together (Romberg).
Sensory loss, a negative symptom, may be missed unless associated with paresthesias or pain. The sensory examination is directed toward demonstrating the type of sensory loss expected (eg, stocking/glove, nerve dermatomal, spinal level). The involved sensory modalities suggest the type of sensory fiber involved (eg, pin/thermal-small fiber, lateral corticospinal, joint position sense–large myelinated fiber, dorsal column). Age-associated decrements of pin, touch, and joint position sense are small. Deep tendon reflexes become less responsive with age, resulting in ankle jerks that can be difficult to elicit.1 Hyperactive reflexes and clonus are atypical in persons over age 75 years and suggest lesions of the corticospinal tracts/neurons. Extensor plantar responses (Babinski signs) are similarly suggestive. Examination of the limbs for tone, coordination, rapid alternating movements (tapping), and tremor are components of the motor exam. Lower-extremity sensory function is an important component of mobility and balance. The evaluation of patients requires medical assessment in addition to a neurologic evaluation. Arthritis can produce a substantial impairment of mobility, although pain and joint dysfunction usually make this diagnosis evident. Frailty is linked to generalized weakness and diminished capacity for aerobic work.
We will now review the more common diseases that affect the anterior horn cell, peripheral nerve, neuromuscular junction, and muscle in the aging population. These diagnoses, which may not be encountered on a frequent or regular basis by the general practitioner or geriatrician, are important to include in the differential diagnosis of an elderly patient with newly acquired weakness in order to effect an appropriate diagnostic work-up and treatment plan. Of course, these diseases are not exclusive to the geriatric population but do either increase in incidence with advancing decades (eg, ALS, inclusion body myositis) or experience a second peak in incidence (eg, myasthenia gravis). Polymyalgia rheumatica, while usually falling under the realm of rheumatology rather than neurology, is briefly reviewed in Part II, as the patient may present with a subjective sense of proximal limb weakness or may not be able to give full effort on muscle strength testing because of limb pain. This is not by any means an exhaustive list of neuromuscular conditions in the elderly, and we would urge readers to also consider hereditary neuropathies with a late presentation, nutritional deficiency neuropathies, neuropathies associated with systemic disease, malignancy or infection, muscular dystrophies with late-onset presentation, toxic myopathies, mitochondrial and metabolic myopathies, or myotonic dystrophy in their differential of the weak elderly patient.
Cervical Spondylotic Radiculomyelopathy
Wear and tear of the cervical spine leads to deterioration of intervertebral disks with subsequent overgrowth of surrounding bone, ligaments, and supporting elements. This excessive tissue growth leads to compression of the spinal cord and/or cervical nerve roots resulting in myelopathy or radiculopathy, which rarely occur together. Radiologic manifestations of cervical spondylosis increase with age, occurring in 10% of persons in the third decade, 50% of those in the fifth decade, and more than 90% of individuals in the seventh decade.2 Despite the presence of prominent radiologic abnormalities, the vast majority of older persons do not have clinical manifestations of the radiculopathy or myelopathy, making it challenging at times to distinguish patients with symptomatic cervical spondylosis from those whose symptoms are due to another process.
Localized neck pain and stiffness are characteristic, although shoulder and upper arm aching or stabbing pain also frequently occur. By contrast, lancinating radicular pain is less common. Slowly progressive gait impairment is the hallmark of the myelopathy but urinary symptoms are unusual. Nonspecific numbness and paresthesias of the upper and/or lower extremities are often encountered, but sensory loss is uncommon or nonspecific (eg, diminished vibration perception at ankles). A narrow-based stiff gait, increased tone, and reflexes with Babinski signs indicate the presence of corticospinal tract dysfunction. The common sites for spondylotic overgrowth that damage the spinal cord are at the C5-6 and C6-7 intervertebral spaces, so motor signs and symptoms may be present in the hands (ie, clumsiness, weakness). If the spondylotic overgrowth has a major lateral component, root compromise may occur, producing weakness and muscle atrophy localized to a nerve root distribution. Narrowing may occur at one or more levels.
Patients with history and examination findings suggestive of spondylotic myelopathy should have a magnetic resonance imaging (MRI) scan, which demonstrates spinal stenosis with cord compression at a site consistent with the clinical picture. An electromyogram (EMG) should be obtained to determine the presence of muscle denervation, which if widespread, suggests the possibility of ALS. Unfortunately, denervation may not be present in early ALS, making this distinction difficult, even in experienced hands. A vitamin B12 level test is mandatory, but other causes of myelopathy, including multiple sclerosis and human T-lymphotropic virus type 1, are unusual at this age, leaving further work-up to the discretion of the clinician.
Spinal cord damage may occur as a result of compression, ischemia, or trauma induced by repetitive neck movement. In patients with spondylotic lesions, forceful neck flexion/extension may produce localized injury to central portions of the cervical spinal cord. This results in prominent hand/arm weakness with a relatively mild myelopathy (ie, lower-extremity weakness, sensory loss). Patients with congenitally narrow spinal canals (eg, achondroplasia) are susceptible to developing the myelopathy. The progression of untreated spondylotic myelopathy is variable, making treatment decisions difficult, with many patients having mild-to-moderate symptoms remaining static or even improving.3
Physical therapy, traction, and a hard cervical collar have been used with modest success. Current surgical techniques are geared toward correcting the mechanism of cord injury with the use of posterior or anterior decompression depending on site(s) of spondylotic damage. Recent surgical series have reported good results, although there have been no treatment trials.2 Patients with a mild myelopathy can be followed. The most compelling scenarios for surgery are a moderate-to-severe deficit with a history of rapid evolution or documented progression of the myelopathy despite best conservative treatment.
Lumbar Spondylosis
The age-related factors generating cervical spondylosis are comparable to those producing lumbar spondylosis. The spinal cord terminates in the conus medullaris (L1), which then become lumbosacral roots (cauda equina) descending through the lumbar spinal canal. Spondylotic narrowing at L4-5 and L3-4 if central compress the canal, and if lateral, a nerve root. Narrowing may occur at one or more levels.
Pseudoclaudication, a characteristic symptom, is uni- or bilateral buttock or lower-extremity pain and/or numbness on walking, which may be relieved by resting, sitting, and bending forward. Progression is slow, and neurologic deficit is only occasionally encountered. Signs of acute radiculopathy (eg, abnormal straight-leg raising) are unusual, although abnormal reflexes and localized weakness are occasionally encountered.4
MRI demonstrates the problem in the vast majority of patients. EMG may be useful in demonstrating root involvement. As in cervical spondylosis, radiologic findings are common in many patients without symptoms, so consistency with the clinical status is important. Ischemia of the lower limbs with true claudication and hip problems are among the diagnoses that should be considered. By contrast with claudication, flexion may relieve the pain of pseudoclaudication, which has been known to occur even with prolonged standing.4 Proposed mechanisms for pseudoclaudication include: hyperextension of the spine, which may increase intervertebral disk protrusion that is relieved by flexion; and ischemia due to compression of radicular arteries during extension, with a similar mechanism of relief by flexion.
Despite the absence of a neurologic deficit, pseudoclaudication may compromise independence and quality of life by interfering with mobility. Pain requiring one or more stops after 50-100 meters of walking may make shopping and social activity difficult. Physical therapy and pain management are of value for getting through periods of exacerbation and may also provide long-term pain relief. In otherwise healthy older persons whose quality of life is being compromised, surgical management should be considered. A recent nonrandomized, as-treated comparison with control of confounding baseline factors comparing surgical (laminectomy) and nonsurgical treatment demonstrated that surgery produced more effective pain relief and improved function.4
Amyotrophic Lateral Sclerosis
ALS is a neurodegenerative disease, the hallmark of which is progressive, painless weakness with both upper and lower motor neuron findings on examination. It is characterized by pure involvement of the motor system with sparing of the oculomotor, sensory, and autonomic systems. Cognition is also usually spared, although there are forms associated with frontotemporal dementia5; one author found that 15% of patients with frontotemporal lobar dementia in a dementia clinic met the criteria for definitive ALS, and a further 30% had signs of possible ALS.6 The incidence is 1-2 per 100,000 per year, and prevalence is 5-7 per 100,000,7 with patients typically manifesting symptoms in their sixth decade.8 However, the age of onset can range from childhood to the ninth decade. Ninety percent of cases are sporadic, and only 10% are familial.9 Mean survival time from onset of symptoms is 32 months and 19 months from time of diagnosis. Death is usually a consequence of respiratory failure within 3-5 years of disease onset. There is a 7% survival rate 5 years after diagnosis.10
The majority of patients present with limb weakness, although 20% of patients start with bulbar signs and symptoms, a presentation that is associated with a worse prognosis. Occasionally, patients’ initial presentation is diaphragmatic and intercostal weakness, manifesting as dyspnea, orthopnea, or even respiratory failure. Symptoms of nocturnal hypoventilation, such as interrupted or poorly restorative sleep, early morning headaches, and excessive daytime somnolence, should be queried, as patients may not volunteer these symptoms.11
Upper motor neuron findings include hypertonicity, hyperreflexia, weakness, and pseudobulbar affect (heightened emotional responses; inappropriate laughing or crying, lability). Lower motor neuron findings include atrophy, hypotonia, cramps, fasciculations, hyporeflexia, weakness, and bulbar dysarthria. Classic ALS may begin with either an upper or lower motor neuron–predominant picture but evolves toward a combination of both upper and lower motor neuron findings. There are rare disease variants that are either pure upper motor neuron in form (primary lateral sclerosis) or pure lower motor neuron (progressive muscular atrophy).
Much of the diagnostic difficulty that arises with ALS stems from the facts that there is no laboratory test that can determine if a patient has the disease, and there are a number of conditions that mimic the disease (eg, cervical radiculomyelopathy, multifocal motor neuropathy, benign cramp fasciculation syndrome).11 The EMG features of fasciculations, fibrillation potentials, and positive sharp waves combined with long duration, high-amplitude motor unit action potentials with a reduced recruitment pattern can be observed in other chronic neurogenic conditions. Compounding this is the fact that patients may present early in the course of their disease, and even though the clinical suspicion of ALS may be raised, there is the understandable reluctance of a clinician to make this diagnosis, given the uniformly fatal outcome in the absence of effective treatments. Once ALS mimics have been excluded, then patients may be stratified into possible, probable, or definite ALS based on the revised El Escorial criteria.12 The benefits of timely diagnosis include prevention of mismanagement of the patient, appropriate treatment, admission to ALS trials before the disease is advanced, psychological benefits for the patient, and early decision making.13
Atrophy and death of motor neurons, along with accumulation of Bunina bodies and Lewy-like inclusions and activation and proliferation of astrocytes and microglia, are seen in ALS.9 The pathogenesis is not understood, which has led to problems in developing effective treatments. Approximately 10% of cases are familial (FALS) and about 10-20% of these are caused by mutations in the SOD1 gene on chromosome 21q22.1 (ALS1). Other genetic loci have been linked to FALS, including 2q33 (ALS2), 20q13.33 (ALS8), 18q21 (ALS3), 16q12 (ALS6), and 20q13 (ALS7).14 However, FALS has a mean age of onset 10 years earlier than sporadic ALS (46 yr vs 56 yr), therefore is less likely to be encountered in the geriatric population. Sporadic ALS (90% of cases) hypotheses include those implicating glutamate excitotoxicity and oxidative stress.
At present there is only one Food and Drug Administration (FDA)–approved drug for the treatment of ALS: riluzole, which is a glutamate release inhibitor. A double-blind, randomized, placebo-controlled trial suggested that it increases length of life, on average, by 3 months in patients with classic ALS.15 Apart from the potential for hepatotoxicity, it is well tolerated. Antioxidant treatments,16 creatine,17 topiramate,18 and recombinant insulin-like nerve growth factor-119 have not been found to have a beneficial effect.
The other thrust of treatment is aimed at symptomatic management, preferably by a multidisciplinary team. Effective palliation exists for symptoms of cramping (quinine, magnesium, carbamazepine), spasticity (baclofen, tizanidine, clonazepam, dantrolene), sialorrhea (glycopyrrolate, benztropine, transdermal hyoscine, amitriptyline, trihexyphenidyl hydrochloride, botulinum toxin type A injection of salivary glands), and pseudobulbar affect (amitriptyline, selective serotonin reuptake inhibitors).11,20
Physical and occupational therapy are required to address a patient’s mobility and self-care needs. Avoidance of falls and subsequent bone fractures is important in maintaining mobility. Speech and language therapy can aid with communication (eg, speech amplification devices). Regular swallow safety assessment and monitoring of body weight are a part of routine care of the patient with ALS. Forced vital capacity (FVC) monitoring is a key part of the clinical assessment, as bilevel positive airway pressure (BiPAP) is usually initiated when FVC is less than 50%.20 In patients with good bulbar function, noninvasive ventilation confers, on average, a 7-month survival benefit, with associated improvement in multiple quality-of-life measures.21 Consideration of percutaneous endoscopic gastrostomy (PEG) tube placement at this juncture (even if bulbar symptoms are not prominent) can avoid difficulties with placement at a later stage, when the FVC is less that 30% and the procedure considered high risk. Invasive ventilation is an option that the majority of patients in Western cultures decline, usually because as respiratory muscles fail, so do their skeletal muscles and thus it becomes increasingly difficult to maintain a reasonable quality of life. Hospice involvement in the latter stages can help these patients achieve a comfortable and dignified death.
Guillain-Barré Syndrome
GBS is a clinical spectrum of self-limited, immune-mediated, acute polyradiculoneuropathies whose hallmark is flaccid, areflexic weakness, usually with sensory (paresthesias and proprioceptive loss) and autonomic (urinary retention, tachycardia, and labile blood pressure) involvement. Patients will frequently report moderate-to-severe back pain as a part of their initial symptomatology.
The incidence is estimated at 1-2 per 100,000 per year,22,23 with a male preponderance and increasing incidence with advancing age.24 However, with very advanced age (> 80 yr) the incidence may decrease.25
Symptoms reach their nadir within 2 weeks in 75% of patients, 3 weeks in 92%, and 4 weeks in 94%26; if the nadir is reached at 8 or more weeks, the disease is classified as chronic inflammatory demyelinating polyradiculoneuropathy (CIDP). There are mild cases in which the patient remains ambulatory throughout the illness. One epidemiological study from the Netherlands found that 28% of patients could walk unaided at their nadir.27 At the opposite end of the spectrum, respiratory failure occurs in one-third of patients with early predictors of mechanical ventilation being early hospital admission (< 7 days from symptom onset), inability to lift the head, and vital capacity of less than 60% predicted.28
Recovery may take weeks or months, with advancing age and greater degree of axonal involvement associated with a worse outcome.22 Half of patients will have a residual large-fiber sensorimotor polyneuropathy,29 but only 6-9% of patients are unable to walk unaided by 1 year.30 Between the years 2000-2004, inpatient mortality rate for GBS in U.S. hospitals was 2.58%.23 Predictors of mortality included endotracheal intubation, older age, comorbidities, cardiac complications, and sepsis. GBS costs the United States an estimated $1.7 billion annually.31
There are a number of inciting factors linked to GBS; use of swine influenza vaccine in 1976-1977 was associated with increased incidence of GBS (1 per 100,000 vaccinations).32 The association of increased incidence of GBS to vaccinations remains unresolved with conflicting reports in the literature.24,33 Antecedent infection, typically upper respiratory or gastrointestinal infection, is reported in 59-70% of cases. Positive serology for Campylobacter jejuni, cytomegalovirus (CMV), Epstein-Barr virus (EBV), or Mycoplasma pneumoniae is more frequently associated with the severe form of the disease (unable to walk at the nadir).30
While the sequence of pathophysiologic events is still being worked out, it is known that macrophages directed against the myelin sheaths strip the axons. One hypothesis, as described by Hughes and Cornblath, is that the “macrophages are targeted to antigens on the surface of the Schwann cells or myelin by activated T lymphocytes.”34 Others postulate that antibody–binding to the Schwann cell, complement fixation, and myelin destruction precede the macrophage invasion.34
Diagnosis of GBS hinges on recognition of the clinical features of the disease, with the physician maintaining a high degree of suspicion in elderly patients with multiple comorbidities. Acute or subacute onset of progressive symmetric limb weakness with distal sensory complaints should awaken the suspicion of this entity in the clinician. Reflexes may be preserved but subsequently lost. Cerebrospinal fluid (CSF) shows albuminocytologic dissociation: if a pleocytosis is found, other diagnoses should be entertained (HIV, lymphoma, Lyme disease). Hyponatremia and elevated transaminases are frequently present. Nerve conduction studies (NCS) are usually performed to further strengthen the case for GBS; a retrospective study of Cleveland Clinic patients undergoing NCS within 7 days of onset of muscle weakness found that the absence of the H reflex, prolonged or absent F-waves, and abnormal upper-extremity sensory nerve action potential combined with normal sural sensory nerve action potentials (“sural sparing”) were characteristic of GBS.35 Testing for antiganglioside antibodies can be performed; the anti-GQ1b antibody is present in 95% of patients with the Miller Fisher GBS variant,36 and patients with the axonal form of GBS are more likely to be seropositive for anti-GD1a antibodies.37
Treatment is largely supportive: attention to deep vein thrombosis prophylaxis, adequate nutrition, aspiration prevention, treatment of concurrent infections, and monitoring for arrhythmias help prevent much of the morbidity associated with GBS. Patients should be monitored for changes in respiratory status, which is frequently done in a critical care setting. Intubation and mechanical ventilation of patients when the vital capacity falls to less than the 15 mL/kg, arterial oxygen tension < 9.3 kPa, or there is inability to protect the airway is based on the Ropper and Kehne criteria.38 Plasmapheresis and intravenous immunoglobulin (IvIG) have come to be part of standard treatment of GBS, and both have proved to be effective in randomized controlled trials in shortening the length of time to recovered ambulation.39,40 Although equally effective,41 there is no additive affect.41 In the anti-GM1 seropositive subgroup, immunoglobulin may be more effective.42 Treatment with IvIG or plasmapheresis does not influence the outcome on Miller Fisher syndrome (ataxia, areflexia, ophthalmoplegia), which may be a reflection of the favorable natural course of this GBS variant.43 The Dutch GBS study group did not observe a significant treatment difference between IvIG plus methylprednisolone versus IvIG alone.44
Acquired Demyelinating Polyneuropathies
Chronic inflammatory demyelinating polyradiculoneuropathy was a term coined by Dyck et al45 in 1982 to describe patients with chronic and progressive as well as relapsing polyradiculoneuropathies. The chronic, progressive form is more frequently encountered in older adults. Most patients have a symmetric, sensorimotor presentation of gradual onset. Paresthesias are frequently reported and less frequently cause pain. Reflexes are diminished or absent, but unlike GBS, dysautonomia is not a feature.
Prevalence is 1-2 per 100,000 population and can be associated with HIV, hepatitis C, diabetes mellitus, osteosclerotic myeloma, and lymphoma.46 CSF protein levels can range from 0.23 g/L to 5.50 g/L.43 Several sets of diagnostic criteria are in existence, but all share a requirement for a combination of the following on electrodiagnostic testing: partial conduction block of a motor nerve; slowing of conduction velocities in two or more motor nerves; prolonged distal motor latencies in two or more nerves; and prolonged or absent F-waves in two or more nerves.46 Nerve biopsy can be considered in patients in whom the diagnosis is strongly suspected but who lack the classic electrophysiologic findings47: the pathologic hallmarks are perivascular inflammatory infiltrates; demyelination; and “onion bulb” formations.
CIDP is an immune-mediated neuropathy and predictably responds to immunosuppressant therapy. Steroids, either daily oral prednisone or pulsed methylprednisolone, are a cornerstone of treatment,45,46,48 in addition to IvIg49,50 and plasmapheresis.51
Distal acquired demyelinating symmetric polyneuropathy (DADS), in which patients present with slowly progressive, distal sensory or sensorimotor symptoms and have characteristic, marked, distal more than proximal, demyelinating features on NCS. Two-thirds of these patients have an IgM-kappa paraproteinemia (DADS-M), and the overwhelming majority are men in their sixth or more decade.52 Anti-myelin–associated glycoprotein antibodies are frequently detected in these patients. Unfortunately, immunosuppressive therapies (prednisone, plasmapheresis, IvIG, interferon-α, and cyclophosphamide) have not been of benefit. There are small series of patients who may have responded to treatment with rituximab.53 The remaining one-third do not have a paraproteinemia (DADS-idiopathic) and may respond to immunosuppression.54
Multifocal motor neuropathy (MMN) with conduction block is a pure motor syndrome and can mimic ALS, as it usually presents with asymmetric, painless, progressive weakness of a distal limb, with a predilection for the upper extremities. Atrophy may occur if there is axonal degeneration secondary to chronic demyelination. Anti-GM1 antibodies are frequently found in these patients but are not specific for the disease and have a sensitivity of 30-50% in MMN with commercial enzyme-linked immunosorbent assay (ELISA) testing.55 The demonstration of conduction block on NCS is not an absolute requirement; if the clinical suspicion is strong, a trial of treatment with IvIg is usually undertaken, as this disease is eminently treatable.54 Steroids and plasmapheresis are not effective and may even exacerbate the condition.
Conclusion
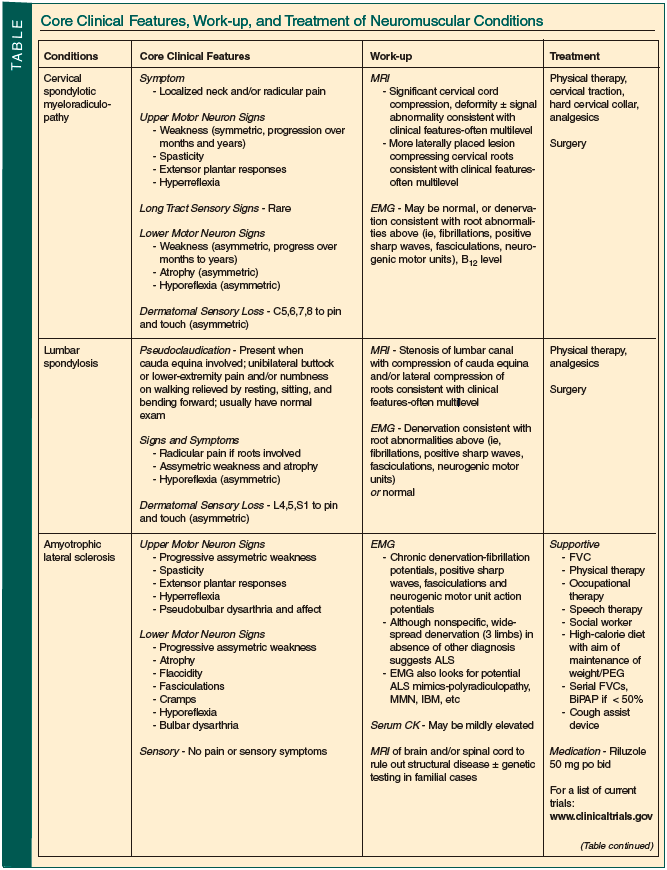
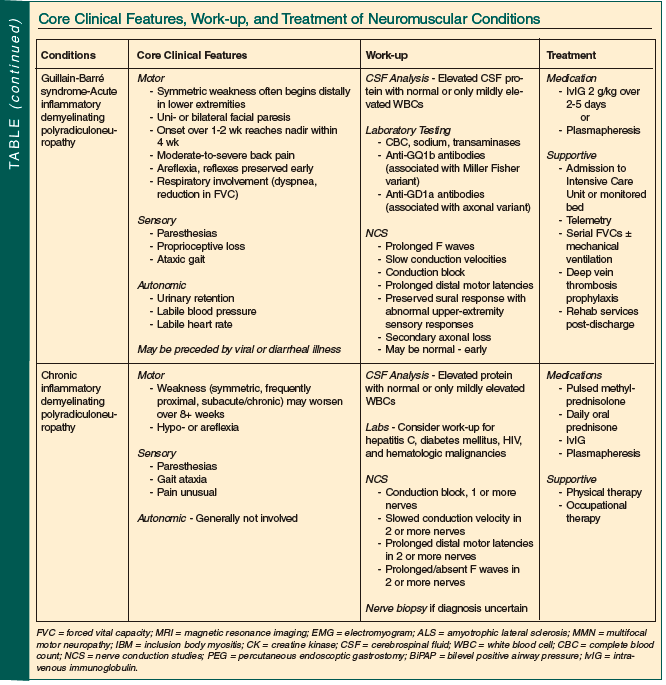
Neuromuscular disease is both common in older persons and often treatable. Although sometimes straightforward, most often diagnosis and management require a neurologist’s input. The Table summarizes the core clinical features, suggested work-up, and usual treatments employed in the conditions discussed.
The authors report no relevant financial relationships.
Dr. Gleeson is from St. Francis Hospital, Hartford, CT; and Dr. Wolfson is Professor and Chair, Department of Neurology, University of Connecticut School of Medicine, Farmington, and Chief of Neurology, Hartford Hospital, Hartford, CT.