
Not Just a Simple Headache
 An 18-year-old male without significant past medical history presented with 2-day history of swelling of the forehead located above the right eye.
An 18-year-old male without significant past medical history presented with 2-day history of swelling of the forehead located above the right eye.
History
The patient reported taking aspirin for the swelling with minimal improvement. He recalled 2 occasions of hitting his forehead beginning 3 months prior, once on a lawn mower handle and again, 1 month later while swimming in a pool.
Laboratory Tests
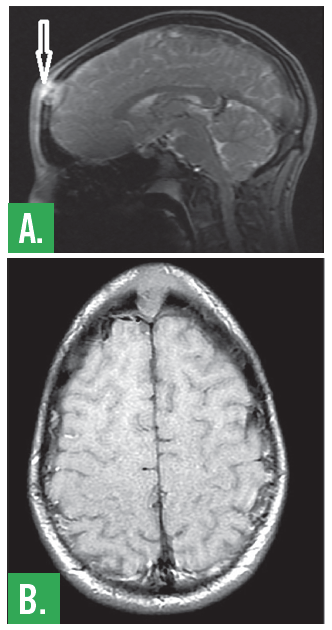
A CT showed a lytic lesion in the midline of the frontal bone involving the inner and outer tables of the calvaria, distinctly separate from the frontal sinus and extending into the epidural and subgaleal space. An MRI was also performed, confirming the presence a 2.1 cm x 1.3 cm x 1.6 cm frontal bone lesion (Figure 1).
Figure 1. MRI of the brain shows solitary lesion of the skull. Central non-enhancing portion extends into the subcutaneous tissues eccentric to the left. There is extensive symmetric frontal subcutaneous enhancement.
Treatment
Due to suspected chronic osteomyelitis of the skull related to his previous head traumas, the patient initially received intravenous ceftriaxone and vancomycin in the emergency department; infectious disease later modified that treatment to a 7-day course of vancomycin and cefepime. Neurosurgery recommended image-guided craniotomy for resection of the skull mass with subgaleal dissection of the tumor, and the patient underwent surgery on hospital day 1. Wound cultures and surgical pathology were taken during the case. Due to the ambiguous pathology of the mass on frozen section, primary cranial reconstruction was not performed at that time.
Preliminary pathology of the mass was consistent with Langerhans cell histiocytosis (LCH) with immunostains confirming CD1a and S100 positivity, and EMA and MITF negativity (Figures 2 and 3). Confirmation of this pathologic diagnosis was made through 2 other expert consultations which confirmed presence of CD1a+, S100+, CD68-, MPO-, EMA-, Vim+, and MITF- Langerhans cells (LC). Both aerobic and anaerobic cultures of the mass were negative for evidence of osteomyelitis.
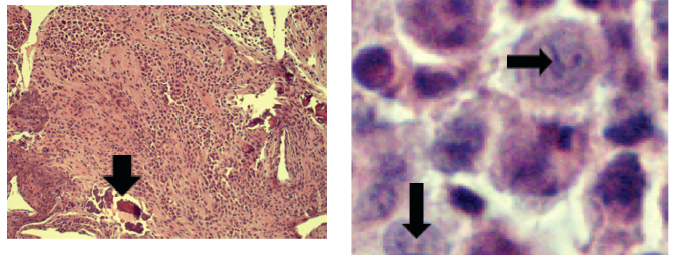
Figure 2. Sheets of polygonal cells have invaded and replaced multiple areas of the Skull bones. Minute fragments of surviving bone are present (arrow).
Figure 3. The tumor cells are consistent with Langerhans histiocytes, which exhibit characteristic “coffee bean” shaped longitudinal grooves (arrows).
Discussion
LCH is a mass proliferation of pathologic LC with indeterminate cells, interdigitating cells and macrophages, macrophages, T-lymphocyte, multinucleated giant histiocytes, and eosinophils.1 The condition was classically subdivided into distinct entities based on clinical manifestations, including Hand-Schüller-Christian disease, Letterer-Siwe disease, and eosinophilic granuloma; these terms have since been abandoned.2 The disease presents most often in children with peak age of diagnosis between 1 and 3 years.3,4 Most cases of LCH are limited to children between the age of 1 and 15 years, although adults can also develop LCH.5,6 There is also a male predilection of 1.5:1.5
In LCH, granulomatous deposits of LC can appear in unifocal (33%) or multifocal (66%) presentations at various sites in the body listed in decreasing frequency: bone (77%), skin (39%), lymph nodes (19%), liver (19%), spleen (13%), oral mucosa (13%), lung (10%) and CNS (6%).7 The initial presentation of LCH is highly variable depending on the organ systems involved. A study of 47 adult patients diagnosed with LCH had a wide variety of presenting symptoms, including skin rash (26%), dyspnea (13%), thirst and polyura (13%), pain (12%), lymphadenopathy and/or splenomegaly (9%), loss of weight (8%), and fever (7%); gum hypertrophy, arthralgia, aural discharge, ataxia and amnesia were also reported.8 Diabetes insipidus is the most common presenting initial sign of LCH in the CNS.9
Definitive diagnosis of LCH requires tissue samples exhibiting both morphologic identification of characteristic LCs with deep-grooved nuclei and immunohistochemical evidence of S100+ and CD1a+.1 Pathognomonic identification of Birbeck granules by electron microscopy is now rarely performed.
Pathophysiology
Nonpathologic LC are antigen-presenting dendritic cells that are present only in the mucosa and skin and play an important role in adaptive immunity.10,11 In the presence of colony-stimulating factors, monocytes with marker GR1 undergo differentiation to become LC.12
The pathogenesis of LCH has been attributed to a dysregulation of immune response to viral infection and aberrant chemokine receptor expression.13 The abnormal retention and proliferation of pathologic LCs is thought to occur through expression of inflammatory cytokines, chemokines, and chemokine receptors. LCH samples from various tissues, including bone, skin, and lymph nodes exhibit aberrant co-expression of CCR6 (normally expressed by immature dendritic cells) and of CCR7 (normally expressed by mature dendritic cells).14,15 LCH samples have also demonstrated the presence of inflammatory chemokines CCL5/RANTES and CXCL11/1-TAC, accounting for the recruitment of eosinophils and CD4+ T-cells in LCH lesions.16 Multifocal manifestations of LCH exhibit dysregulation expression of the cell adhesion molecule E-cadherin suggesting that the loss of this factor contributes to disseminated disease.17 The immunostimulatory function of normal activated LC is also exhibited by acidic organelles, known as Birbeck granules, that process antigens for T-cell sensitization.18 The lack of these granules seen in normal resting LC and the characteristic presence of these granules in LCH further confirms an immunomodulatory component to the pathogenesis of LCH.
LCH is also associated with somatic mutations in the BRAF gene and subsequent activation of the Ras-Raf oncogenic pathway. Studies demonstrate that the majority of samples of pathologic dendritic cells in LCH exhibit BRAF-V600E gene mutation.19,20 Oncogenic BRAF mutations are associated with subsequent activation of Ras-Raf pathway, including tyrosine kinases MEK and ERK.21 Molecular studies also confirm the expression of activated phospho-MEK and phospho-ERK in LCH does not vary with BRAF mutational status which confirms that LCH is a clonally derived neoplasm, although non-BRAF inciting mutations are not known at this time.20 Similar to the understood pathophysiology of Hodgkin lymphoma, LCH is therefore recognized as a neoplastic disease associated with intense expression of inflammatory cytokines and chemokines.22
Risk Stratification, Treatment, and Prognosis
The treatment for LCH is determined by the extent and nature of the patient’s disease. Guidelines from the Histiocyte Society base risk stratification on the presence of disseminated disease (unifocal vs multifocal disease) and the presence of organ dysfunction.2 In the presence of organ dysfunction, risk stratification is further based on the type of organ involvement. High-risk disease is associated with pulmonary, liver, spleen, and hematopoietic involvement; low-risk disease is associated with skin, bone, lymph node, and pituitary involvement.2 Due to evidence of high 5-year survival in pediatric-onset pulmonary involvement of LCH, it is suggested that it be no longer considered a high-risk organ.23
Cutaneous LCH is common, however, limited (unifocal) cutaneous disease is very rare and is likely represents inadequately evaluation patients at presentation.24 Children with presumed isolated cutaneous LCH can be monitored, however, 40% of these patients have or will develop multisystem disease.24,25 Symptomatic patients with skin-limited LCH should be treated with oral methotrexate and 6-mercaptopurine adjusted for myelosuppression.26
Patients with unifocal bony lesions, such as our patient, can be treated effectively with resection and/or corticosteroid injection.27 These lesions only have 10% recurrence rate once treated.28 However, bony LCH lesions in the craniofacial region (ie, orbital, mastoid, temporal, sphenoid, zygomatic, ethmoid, maxilla, paranasal sinuses, or anterior or posterior cranial fossa) are considered high risk for CNS involvement.7 Craniofacial LCH has been identified as a risk factor for developing diabetes insipidus secondary to LCH involving the posterior pituitary gland, which increases with extent of disease and recurrence.7
Multifocal disease involving only low-risk organs is treated with vinblastine and prednisone for 1 year. Multifocal disease involving high-risk organs is treated with vinblastine, prednisolone, and mercaptopurine for a period of 6 weeks, followed by response-triggered milder continuation therapy for a total 1-year duration of therapy. The LCH-III trial found that lower recurrence rates are achieved by prolonged (1 year) therapy in patients with both low- and high-risk organ involvement.29 Recurrence is common, roughly around 40%, among those with multifocal disease and is independent of high/low risk organ involvement at initial presentation.30 Age is no longer considered an important prognostic factor in patients <2 years old, as the LCH-II study demonstrated that these patients without high-risk organ involvement had similar mortality to older patients.31
Treatment options for CNS LCH and craniofacial LCH high risk for CNS involvement include cytarabine, vinblastine/prednisone, cladribine, clofarabine, and resection with or without low-dose radiation therapy.32-35
Outcome of the Case
The remainder of the patient’s hospital course was unremarkable and the patient was discharged on hospital day 7. At the time of discharge, the patient had complained of swollen left sided submental lymph nodes and bilateral preauricular lymph nodes bilaterally which resolved on outpatient follow-up 2 weeks post-discharge. The patient returned for cranioplasty 2 months following the initial craniotomy and mass excision.
Bensson Samuel, MD, Pg Dip, is an internal medicine resident at St. Luke's University and Health Network in Bethlehem, PA.
Gloria Fioravanti, DO, is the internal medicine program director at St. Luke’s University Hospital and Health Network and an associate professor of medicine at Temple University School of Medicine, both in Bethlehem, PA.
Jennifer Axelband, DO, is a critical care attending at St. Luke’s University Hospital and Health Network and an associate clinical professor at Temple University School of Medicine, both in Bethlehem, PA.
Alden Smith, BA, is a medical student at Temple University School of Medicine in Bethlehem, PA.
Michael Hughes, BS, is a medical student at Temple University School of Medicine in Philadelphia, PA.
Santo Longo, MD, is the emeritus chief of pathology at St.Luke’s University Health Network in Bethlehem, PA.
Emily Keeler, DO, is an internal medicine resident at St.Luke’s University Hospital and Health Network in Bethlehem, PA.
References:
1.Arico, M, Clementi R, Caselli D, Danesino C. Histiocytic disorders. Hematol J. 2003;4(3):
171-179.
2.Satter EK, High WA. Langerhans cell histiocytosis: a review of the current recommendations of the Histiocyte Society. Pediatr Dermatol. 2008;25(3):291-295.
3.Carstensen H, Ornvold K. The epidemiology of LCH in children in Denmark, 1975-1989 [abstract]. Med Pediatr Oncol. 1993;
21(5):387.
4. Guyot-Goubin A, Donadieu J, Barkaoui M, et al. Descriptive epidemiology of childhood Langerhans cell histiocytosis in France, 2000-2004. Pediatr Blood Cancer. 2008;51(1):71-75.
5.Salotti JA, Nanduri V, Perace MS, et al. Incidence and clinical features of Langerhans cell histiocytosis in the UK and Ireland. Arch Dis Child. 2009;94(5):376-380.
6.Baumgartner I, von Hochstetter A, Baumert B, et al. Langerhans'-cell histiocytosis in adults. Med Pediatr Oncol. 1997;28(1):9-14.
7.Grois N, Potschger U, Prosch H, et al. Risk factors for diabetes insipidus in langerhans cell histiocytosis. Pediatr Blood Cancer. 2006;46(2):228-233.
8.Malpas JS, Norton AJ. Langerhans cell histiocytosis in the adult. Med Pediatr Oncol. 1996;27(6):540-546.
9.Dunger DB, Broadbent V, Yeoman E, et al. The frequency and natural history of diabetes insipidus in children with Langerhans-cell histiocytosis. N Engl J Med. 1989;321(17):1157-1162.
10. Sparber F. Langerhans cells: an update. J Dtsch Dermatol Ges. 2014;12(12):1107-1111.
11. Teunissen MB. Dynamic nature and function of epidermal Langerhans cells in vivo and in vitro: a review, with emphasis on human Langerhans cells. Histochem J. 1992;24(10):697-716.
12. Ginhoux F, Tackle F, Angeli V, et al. Langerhans cells arise from monocytes in vivo. Nat Immunol. 2006;7(3):265-273.
13. Degar BA, Rollins BJ. Langerhans cell histiocytosis: malignancy or inflammatory disorder doing a great job of imitating one? Dis Model Mech. 2009;2(9-10):436-439.
14. Garabedian L, Struyf S, Opdenakker G, et al. Langerhans cell histiocytosis: a cytokine/chemokine-mediated disorder? Eur Cytokine Netw. 2011;22(3):148-153.
15. Fleming MD, Pinkus JL, Fournier MV, et al. Coincident expression of the chemokine receptors CCR6 and CCR7 by pathologic Langerhans cells in Langerhans cell histiocytosis. Blood. 2003;101(7): 2473-2475.
16. Annels NE, Da Costa CE, Prins FA, et al. Aberrant chemokine receptor expression and chemokine production by Langerhans cells underlies the pathogenesis of Langerhans cell histiocytosis. J Exp Med. 2003;197(10):1385-1390.
17. Geissmann F, Emile JF, Andry P, et al. Lack of expression of E-cadherin is associated with dissemination of Langerhans' cell histiocytosis and poor outcome. J Pathol. 1997;181(3):301-304.
18. Stossel H, Koch F, Kampgen E, et al. Disappearance of certain acidic organelles (endosomes and Langerhans cell granules) accompanies loss of antigen processing capacity upon culture of epidermal Langerhans cells. J Exp Med. 1990;172(5):1471-1482.
19. Berres ML, Lim KP, Peters T, et al. BRAF-V600E expression in precursor versus differentiated dendritic cells definesclinically distinct LCH risk groups. J Exp Med. 2014;211(4):669-683.
20.Badalian-Very G, Vergilio JA, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116(11):
1919-1923.
21.Montagut C, Settleman J. Targeting the RAF-MEK-ERK pathway in cancer therapy. Cancer Lett. 2009;283(2):125-134.
22. Badalian-Very G, Vergilio JA, Fleming M, et al. Pathogenesis of Langerhans cell histiocytosis. Annu Rev Pathol. 2013;8:1-20.
23. Ronceray L, Potschger U, Janka G, et al. Pulmonary involvement in pediatric-onset multisystem Langerhans cell histiocytosis: effect on course and outcome. J Pediatr. 2012;161(1):129-133.
24. Simko SJ, Garmezy B, Abhyankar H, et al. Differentiating skin-limited and multisystem Langerhans cell histiocytosis. J Pediatr. 2014;
165(5):990-996.
25. Lau L, Krafchik B, Trebo MM, Weitzman S. Cutaneous Langerhans cell histiocytosis in children under one year. Pediatr Blood Cancer. 2006;46(1):66-71.
26. Allen CE, Ladisch S, McClain KL. How we treat Langerhans cell histiocytosis. Blood. 2015.
27. Nauert C, Zornoza J, Ayala A, Harle TS. Eosinophilic granuloma of bone: diagnosis and management. Skeletal Radiol. 1983;10(4):
227-235.
28. Jubran RF, Marachelian A, Dorey F, Malogolowkin M. Predictors of outcome in children with Langerhans cell histiocytosis. Pediatr Blood Cancer. 2005;45(1):37-42.
29. Gadner H, Minkov M, Grois N, et al. Therapy prolongation improves outcome in multisystem Langerhans cell histiocytosis. Blood. 2013;121(25):5006-5014.
30. Minkov M, Steiner M, Potschger U, et al. Reactivations in multisystem Langerhans cell histiocytosis: data of the international LCH registry. J Pediatr. 2008;153(5):700-705.
31. Gadner H, Grois N, Potschger U, et al. Improved outcome in multisystem Langerhans cell histiocytosis is associated with therapy intensification. Blood. 2008;111(5):2556-2562.
32. Allen CE, Flores R, Rauch R, et al. Neurodegenerative central nervous system Langerhans cell histiocytosis and coincident hydrocephalus treated with vincristine/cytosine arabinoside. Pediatr Blood Cancer. 2010;54(3):416-423.
33. Simko SJ, Tran HD, Jones J, et al. Clofarabine salvage therapy in refractory multifocal histiocytic disorders, including Langerhans cell histiocytosis, juvenile xanthogranuloma and Rosai-Dorfman disease. Pediatr Blood Cancer. 2014;61(3):479-487.
34. Dhall G, Finlay JL, Dunkel IJ, et al. Analysis of outcome for patients with mass lesions of the central nervous system due to Langerhans cell histiocytosis treated with 2-chlorodeoxyadenosine. Pediatr Blood Cancer. 2008;50(1):72-79.
35. Kasper EM, Aguirre-Padilla DH, Alter RY, et al. Histiocytosis X: characteristics, behavior, and treatments as illustrated in a case series. Surg Neurol Int. 2011;2:57.