
Young Boy With Symmetric Muscle Weakness and Rash
What's Your Diagnosis?
Sharpen Your Diagnostic Skills
HISTORY
 A 5-year-old white boy had fever (temperature, up to 40.5ºC [105ºF]), progressive weakness of the lower extremities, and weight loss of about 3 kg over the past 3 weeks. During this time, he also had decreased appetite, dull abdominal pain, persistent fatigue, a nasal quality of speech, and an erythematous rash on the face, neck, and abdomen. For the past 10 days, he had bilateral leg and thigh pain and difficulty in rising from a sitting position. Recently, he had refused to walk. Four weeks earlier, he had undergone tonsillectomy and adenoidectomy, with bilateral myringotomy and tube placement.
A 5-year-old white boy had fever (temperature, up to 40.5ºC [105ºF]), progressive weakness of the lower extremities, and weight loss of about 3 kg over the past 3 weeks. During this time, he also had decreased appetite, dull abdominal pain, persistent fatigue, a nasal quality of speech, and an erythematous rash on the face, neck, and abdomen. For the past 10 days, he had bilateral leg and thigh pain and difficulty in rising from a sitting position. Recently, he had refused to walk. Four weeks earlier, he had undergone tonsillectomy and adenoidectomy, with bilateral myringotomy and tube placement.
Results of a laboratory workup (including rheumatoid factor, Epstein-Barr and parvovirus studies, hepa-titis panels, HIV test, complete blood cell count, and blood culture) obtained within past 3 weeks were normal. Immunizations were up-to-date. Child had been given acetaminophen and ibuprofen, as needed. No known drug or food allergies. Diet was well-rounded, with no restrictions.
No family history of oncologic, rheumatologic, or immunosuppressive diseases. Age-appropriate developmental milestones had been met. No recent travel or exposure to sick contacts or animals. Child lived with mother, father, and 2 siblings, all of whom were healthy.
PHYSICAL EXAMINATION
Patient mildly pale, visibly tired. Height, 110 cm (50th percentile); weight, 16 kg (25th
percentile). Temperature, 37.7ºC (99.8ºF); heart rate, 111 beats per minute; respiration rate,
28 breaths per minute; blood pressure, 104/78 mm Hg. Bilateral periorbital edema with a confluent macular erythematous rash, especially prominent on the upper eyelids, extended across the cheeks and nasal bridge, spared the nasolabial folds (like the rash in the child shown). Child had similar rash on the posterior neck and periumbilical regions, along the waistline. No petechial or purpuric lesions or skin findings on fingers or nailbeds.
Palpation of the anterior thighs revealed exquisite bilateral tenderness and weakness of the proximal muscles, including abdominal and hip flexors. Child unable to arise from seated position without assistance. Neck flexors moderately weakened; extensors intact. Full range of motion of all joints, without redness, swelling, pain, or warmth. Gait limited, secondary to pain, weakness.
Abdominal findings normal. No heart murmurs. Reflexes normal. Cranial nerves intact.
WHAT'S YOUR DIAGNOSIS?
What's Your Diagnosis?
ANSWER: JUVINILE DERMATOMYOSITIS
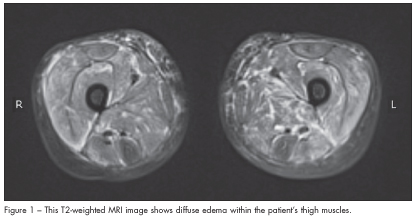
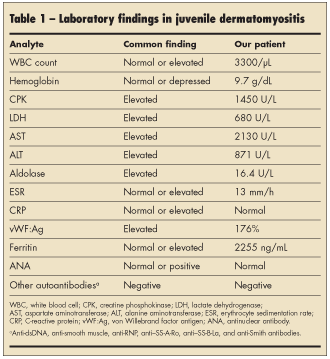 After an extensive laboratory workup, the patient was found to have mild pancytopenia and significantly elevated muscle enzyme levels (Table 1). All other laboratory values, including g-glutamyltransferase, uric acid, prothrombin time, partial thromboplastin time, and a chemistry panel, were normal. The abnormal laboratory results coupled with the muscle pain and weakness prompted an MRI evaluation of the pelvis and lower extremities. Findings revealed extensive myositis and subcutaneous edema, consistent with dermatomyositis (Figure 1).
After an extensive laboratory workup, the patient was found to have mild pancytopenia and significantly elevated muscle enzyme levels (Table 1). All other laboratory values, including g-glutamyltransferase, uric acid, prothrombin time, partial thromboplastin time, and a chemistry panel, were normal. The abnormal laboratory results coupled with the muscle pain and weakness prompted an MRI evaluation of the pelvis and lower extremities. Findings revealed extensive myositis and subcutaneous edema, consistent with dermatomyositis (Figure 1).
INCIDENCE AND ETIOLOGY
Juvenile dermatomyositis (JDM), a rare autoimmune disorder, is the most common inflammatory myopathy of childhood; 3.2 cases per million are reported annually.1,2 It is primarily a vasculopathy of muscular and dermal capillaries and results in various clinical sequelae. Although the cause is not fully understood, a genetic predisposition (involving B8, DRB1*0301, and DQA1*0501 HLA alleles) and environmental factors (recent bacterial or viral infection and excessive sun exposure) appear to play a role in triggering the inflammatory response within affected tissue.1,2
The peak incidence is highest among white girls, with an average age at onset of 7 years. Most cases pre-sent in the spring and summer months, which suggests seasonal variability.3 Our patient presented in May.
AND RADIOGRAPHIC FINDINGS
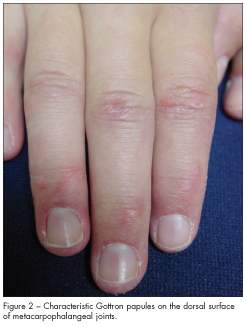 The appearance of a rash on sun-exposed areas of the body (possibly precipitated by sunlight) usually prompts consideration of JDM. An erythematous, violaceous periorbital heliotrope rash (as shown on page 239) may extend along the bridge of the nose and the malar aspects of the face. Scattered telangiectasias may be present within the rash. Edema is frequently noted periorbitally; however, extension to the scalp has been described.5 Gottron papules—scaly, erythematous, papular lesions on the dorsal surface of the knuckles—are also common (Figure 2), although they were absent in our patient. A similar rash may develop on the extensor surfaces of the knees, elbows, and ankles. Nailfold changes, dystrophic calcinosis, and skin ulcerations are other skin findings that correspond to the severity and extent of the disease.
The appearance of a rash on sun-exposed areas of the body (possibly precipitated by sunlight) usually prompts consideration of JDM. An erythematous, violaceous periorbital heliotrope rash (as shown on page 239) may extend along the bridge of the nose and the malar aspects of the face. Scattered telangiectasias may be present within the rash. Edema is frequently noted periorbitally; however, extension to the scalp has been described.5 Gottron papules—scaly, erythematous, papular lesions on the dorsal surface of the knuckles—are also common (Figure 2), although they were absent in our patient. A similar rash may develop on the extensor surfaces of the knees, elbows, and ankles. Nailfold changes, dystrophic calcinosis, and skin ulcerations are other skin findings that correspond to the severity and extent of the disease.
Symmetric proximal muscle weakness is often a slow, progressive process in JDM. The weakness may be apparent not only in the extremities but also in the neck and trunk, as in this case. As in other muscular dystrophies, common activities, such as arising from the floor, climbing stairs, and walking, may be significantly impaired. Patients may exhibit the Gower sign—the need to use the hands and arms to push oneself up from a squatting position.
Other systems that may be affected in JDM include the eye (vision loss), heart (conduction disturbances), abdomen (constipation, abdominal pain, GI bleeding), esophagus (dysphagia), and pharynx (dysphonia).
MANAGEMENT
The mainstay of treatment of JDM is systemic corticosteroids, oral or intravenous.6 Treatment regimens vary among institutions and are often tailored in coordination with a pediatric rheumatologist, according to the severity of disease (Table 2). Additional medical therapies include immunosuppressive agents (methotrexate, cyclosporin, and azathioprine), which are used primarily in corticosteroid-refractory or toxic cases and in relapses.7-11 The use of high-dose intravenous immunoglobulin (IVIG) in JDM is controversial, although it is thought to be useful in
corticosteroid-resistant cases.12 Hydroxychloroquine is another therapeutic adjunct thought to be most effective in patients with prominent cutaneous findings.7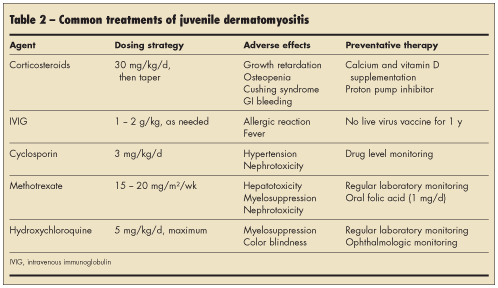
Therapy is continued until clinical signs and laboratory values normalize. Children also require calcium and vitamin D supplementation to minimize the effects of high-dose corticosteroid therapy on bone growth.13
OUTCOME IN THIS CASE
Our patient initially received pulse intravenous methylprednisolone (30 mg/kg/d) for 3 consecutive days in the hospital; this was reduced to weekly boluses as an outpatient. In addition, oral prednisone (2 mg/kg/d), methotrexate (15 mg/m²/wk), and folate supplementation (1 mg/d) was instituted in an attempt to minimize the long-term cumulative corticosteroid use. Daily hydroxychloroquine (5 mg/kg/d) was started, and IVIG (2 g/kg) was administered before discharge because muscle enzyme levels remained elevated after initial treatment. He was also given a proton-pump inhibitor (to minimize gastric bleeding) and calcium and vitamin D supplementation.
After 2 months of treatment, the muscle enzyme levels had normalized, as did the muscle strength in the neck, abdomen, and lower extremities. At the patient’s last clinic visit, he had some mild cutaneous involvement of the face and neck. He continues to receive monthly pulse intravenous methylprednisolone and IVIG. The oral prednisone regimen is currently being weaned. ■
REFERENCES:
1. Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet. 2003;362:971-982.
2. Feldman BM, Rider LG, Reed AM, Pachman LM. Juvenile dermatomyositis and other idiopathic inflammatory myopathies of childhood. Lancet. 2008;371:2201-2212.
3. Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med.1975;292:344-347.
4. Keim DR, Hernandez RJ, Sullivan DB. Serial magnetic resonance imaging in juvenile dermatomyositis. Arthritis Rheum. 1991;34:1580-1584.
5. Kasteler JS, Callen JP. Scalp involvement in dermatomyositis. Often overlooked or misdiagnosed. JAMA. 1994;272:1939-1941.
6. Laxer RM, Stein LD, Petty RE. Intravenous pulse methylprednisolone treatment of juvenile dermatomyositis. Arthritis Rheum. 1987;30:328-324.
7. Ramanan AV, Campbell-Webster N, Ota S, et al. The effectiveness of treating juvenile dermatomyositis with methotrexate and aggressively tapered corticosteroids. Arthritis Rheum.2005;52:3570-3578.
8. Niakan E, Pitner SE, Whitaker JN, Bertorini TE. Immunosuppressive agents in corticosteroid-refractory childhood dermatomyositis. Neurology. 1980;30:286-291.
9. Jacobs JC. Methotrexate and azathioprine treatment of childhood dermatomyositis.Pediatrics.1977;59:212-218.
10. Miller LC, Sisson BA, Tucker LB, et al. Methotrexate treatment of recalcitrant childhood dermatomyositis. Arthritis Rheum. 1992;35:1143-1149.
11. Heckmatt J, Hasson N, Saunders C, et al. Cyclosporin in juvenile dermatomyositis. Lancet.1989;1:1063-1066.
12. Manlhiot C, Tyrrell PN, Liang L, et al. Safety of intravenous immunoglobulin in the treatment of juvenile dermatomyositis: adverse reactions are associated with immunoglobulin A content.Pediatrics. 2008;121:e626-e630.
13. Warady BD, Lindsley CB, Robinson FG, Lukert BP. Effects of nutritional supplementation on bone mineral status of children with rheumatic diseases sreceiving corticosteroid therapy. J Rheumatol. 1994;21:530-535.