
Peer Reviewed
An Infant Girl With Persistent Hyponatremia: What’s the Cause?
A 7-month old girl presented to the Emergency Department with vomiting, fever, and lethargy. Three days before presentation, she developed increasingly frequent nonbilious, nonbloody emesis. This was soon followed by a low-grade fever of 37.8°C and progressively decreasing oral intake and urine output, with only 2 wet diapers in the last 24 hours. The patient’s mother and several other children under the care of the patient’s babysitter had all recently had a self-limited illness with similar symptoms.
The infant’s past medical history was notable for poor weight gain. She was born at term weighing 3.4 kg (70th percentile), but began falling off the growth curve around 1 month of age. Her appetite and interest in feeding was excellent, but she vomited frequently and in large amounts, typically immediately after feeding. Though she was initially exclusively breastfed, she was ultimately switched to a hypoallergenic formula. Six weeks before her presentation, she was evaluated by a pediatric gastroenterologist and treated with ranitidine for gastroesophageal reflux and lactulose for constipation.
In the Emergency Department, the infant’s weight was 5 kg (<1st percentile) with a length of 61 cm (1st percentile) and head circumference of 41 cm (5th percentile). Her temperature was 37.3°C, pulse was 182 beats per minute, and respiratory rate was 32 breaths/min. Physical examination showed that the infant was ill-appearing and undernourished, with decreased subcutaneous fat. She had nondysmorphic facial features and dry mucous membranes. Her anterior fontanelle was slightly depressed. She was tachycardic, with delayed capillary refill (4 seconds) but warm extremities. Her lungs were clear to auscultation, and though she was tachypneic, she had no increased work of breathing. Her abdomen was soft, nontender, and without organomegaly. Bowel sounds were normoactive. She cried weakly when examined and sucked weakly on a bottle when it was offered.
A laboratory evaluation was performed. A complete blood count was within normal limits and revealed a hemoglobin of 12.0 g/dL, hematocrit of 38.8%, white blood count of 9800 /μL, and platelets of 186 × 103 /μL. A chemistry panel demonstrated a serum sodium of 164 mEq/L (reference range 133-142 mEq/L), chloride of 126 mEq/L (reference range 96-108 mEq/L), blood urea nitrogen of 20 mg/dL (reference range 2-19 mg/dL), glucose of 48 mg/dL(reference range 60-90 mg/dL) and normal values for total carbon dioxide, calcium, creatinine and potassium. Measurements of transaminases, creatinine phosphokinase, cortisol, and ammonia were all within normal limits. Urinalysis results showed a urine pH of 5.5, urine specific gravity of 1.020, and no abnormalities by dipstick.
The patient was given 5 mL/kg of 10% dextrose in water to correct hypoglycemia and 20 mL/kg of 0.9% sodium chloride intravenously to address volume depletion. After receiving these fluids, her glucose was 82 mg/dL, and her mental status and general physical appearance were improved. She was then admitted to the general pediatric inpatient service for further evaluation, monitoring, and correction of electrolyte abnormalities.
The initial diagnostic impression was that the patient’s hypernatremia was secondary to dehydration and inadequate free water intake in the context of a viral illness. Therefore, the patient was started on 5% dextrose with 0.45% sodium chloride intravenously at 31 mL/h (approximately 1.5 times the Holliday-Segar maintenance rate). Though the patient’s urine output improved to 4 mL/kg/h on the first night of hospitalization, her serum sodium increased to 170 mEq/L. Further increases in the prescribed fluid during the first hospital day led to additional increases in urine output without significant decreases in the serum sodium.
As a result of persistent hypernatremia, the patient’s serum and urine osmolality were evaluated. The serum osmolality was 337 mOsm/kg (reference range: 275-295 mOsm/kg) and urine osmolality was 184 mOsm/kg, much lower than expected in the context of serum hyperosmolarity. The production of a dilute urine in the face of hyperosmolality is physiologically inappropriate, so a diagnosis of diabetes insipidus was made.
Discussion
Diabetes insipidus refers to the production of excessive amounts of dilute urine. The name is derived in part from the Latin for “tasteless,” and was first used to by 18th century physicians to distinguish patients with polyuria and watery urine from those with sweet-tasting urine (diabetes mellitus).1 Because urine concentration occurs in the distal tubule under the action of antidiuretic hormone (ADH), diabetes insipidus is caused by either reduced production of ADH (central diabetes insipidus) or by failure of the kidney to respond to ADH (nephrogenic diabetes insipidus). Providing a physiologic dose of desmopressin, a synthetic form of ADH, is used to distinguish between these 2 possibilities. A simple algorithm for evaluating patients with hypernatremia is shown in Figure 1.
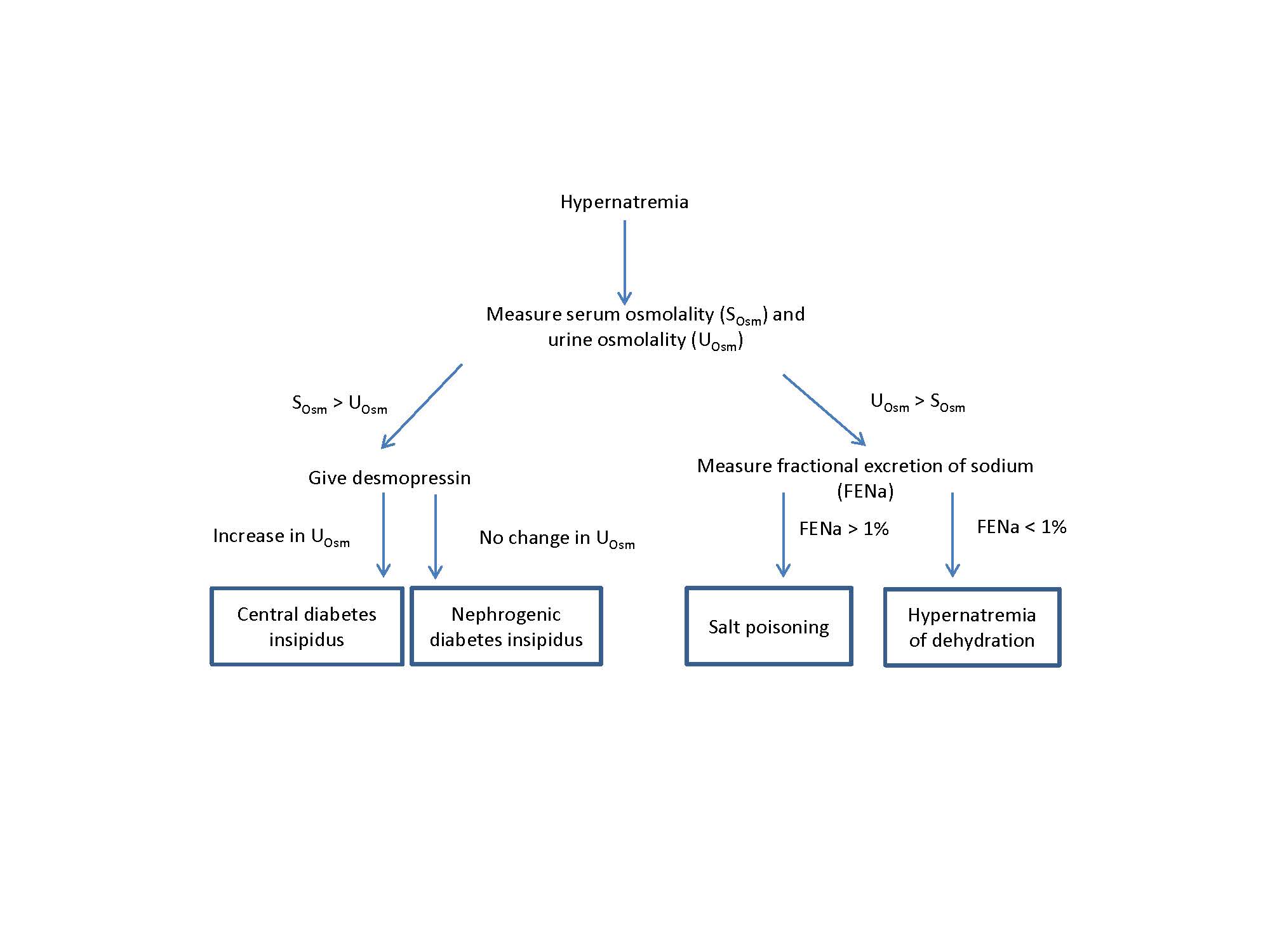
In accordance with this algorithm, the patient received a dose of desmopressin acetate. There was no effect on her serum sodium, urine osmolality, or urine volume, establishing the final diagnosis of nephrogenic diabetes insipidus. She was transferred to the nephrology service for electrolyte and caloric management.
Although nephrogenic diabetes insipidus can be acquired (particularly following chronic therapy with lithium),2 most patients with the condition have an identifiable genetic mutation. In approximately 90% of cases, nephrogenic diabetes insipidus is inherited as an X-linked recessive loss-of-function mutation of the AVPR2 gene encoding the arginine vasopressin type 2 receptor expressed in the kidney tubule. Before the availability of molecular genetic testing, careful study of family pedigrees suggested that most nephrogenic diabetes insipidus patients in North America could be traced to common ancestors on the Hopewell, a ship that brought a group of Ulster Scot immigrants to Halifax, Nova Scotia in 1761. Interestingly, historical records from the early settlers in Nova Scotia document the existence of a folktale, known as the “water drinker’s curse,” to explain the existence of nephrogenic diabetes insipidus in their community. According to the legend, because a Scots-Irish mother refused to allow a gypsy mother and her thirsty son to drink from their well, a curse was cast on the family, and henceforth some boys were afflicted with an insatiable thirst.3
Less commonly, nephrogenic diabetes insipidus can be caused by autosomal dominant or recessive mutations in the APQ2 gene, which encodes the aquaporin-2 channels that allow water molecules to pass across the tubular epithelium.4 The gender of the patient meant that the nephrogenic diabetes insipidus may have been an aquaporin mutation, a suspicion that was subsequently proven accurate by genetic sequencing.
The most prominent features of nephrogenic diabetes insipidus are polyuria and polydipsia, though these signs may be difficult to elicit specifically in an infant. Patients may also report dizziness, weakness, nocturia, fatigue, irritability, and constipation. Failure to thrive and growth retardation are common, and many patients may present with severe dehydration following even minor illnesses.
Unfortunately, children with nephrogenic diabetes insipidus who have frequent episodes of dehydration have a high risk for developing mental retardation.4 The serum hyperosmolality that occurs in severe hypernatremia induces cellular brain shrinkage, which in turn can cause seizures or vascular rupture, resulting in cerebral bleeding, subarachnoid hemorrhage, or permanent neurologic damage and death.
The brain adapts to chronic hypernatremia, which means that rapid normalization of the serum sodium can be damaging, as fluid moves rapidly into the brain cells causing cerebral edema, coma, convulsions, and death. Unless the duration of hypernatremia is known to be less than 24 hours, hypernatremia should be corrected slowly (no faster than 0.5-1.0 mEq/L per hour) to allow the brain to reacclimate to a lower serum osmolality.5
The ultimate goal for treating nephrogenic diabetes insipidus is to maintain fluid and electrolyte homeostasis by adequately replacing urinary losses of water through appropriate water intake. In patients that are very polyuric, this approach may first require therapeutic maneuvers to reduce the urine volume. As an initial step, the infant’s formula was changed to Similac® PM 60/40 to reduce the potential renal solute load.6 The renal solute load represents the mineral content of the formula requiring excretion by the kidney, including urea, sodium, potassium, chloride, and phosphorus. Excessive loss of urine solutes increases urinary fluid loss as well. Formula was administered via nasogastric feeding tube, with additional free water added via a Y tubing system and adjusted appropriately to correct the serum sodium in a controlled fashion.
Even with a low solute formula, the patient remained very polyuric, producing >1.5 L of dilute urine daily. She was unable to consume oral feedings in a volume sufficient to maintain a normal serum sodium due to frequent emesis and gastroesophageal reflux. Therefore, to decrease the urine output, and the fluid intake required, the infant was started on both a thiazide (hydrochlorothiazide, 1 mg/kg twice daily) and a potassium-sparing diuretic (amiloride, 0.1 mg/kg three times daily).
Though it is initially somewhat counterintuitive to prescribe diuretics to a patient with polyuria, the initial hypovolemia caused by thiazide diuretics leads to increased reabsorption of water from the proximal tubule. Potassium-sparing diuretics potentiate this effect and limit the urinary potassium wasting. Following the addition of diuretics, the patient’s urine output decreased to approximately 1200 mL daily, her serum sodium stabilized in the low normal range, and she began to gain weight steadily. At outpatient follow-up visits, she has continued to show excellent catch up growth and normal serum sodium levels, with no further admissions to the hospital.
1. Eknoyan G. A history of diabetes insipidus: paving the road to internal water balance. Am J Kidney Dis. 2010;56(6):1175-1183.
2. Garofeanu CG, Weir M, Rosas-Arellano MP, Henson G, Garg AX, Clark WF. Causes of reversible nephrogenic diabetes insipidus: a systematic review. Am J Kidney Dis. 2005;45(4):626-637.
3. Bode HH, Crawford JD. Nephrogenic diabetes insipidus in North America – the Hopewell hypothesis. N Engl J Med. 1969;280(14):750-754.
4. Bockenhauer D, Bichet DG. Pathophysiology, diagnosis and management of nephrogenic diabetes insipidus. Nat Rev Nephrol. 2015;11(10):576-588.
5. Adrogué HJ, Madias NE. Hypernatremia. N Engl J Med. 2000;342(20):1493-1499.
6. Ziegler EE, Fomon SJ. Potential renal solute load of infant formulas. J Nutr. 1989;119(12 Suppl):1785-1788.